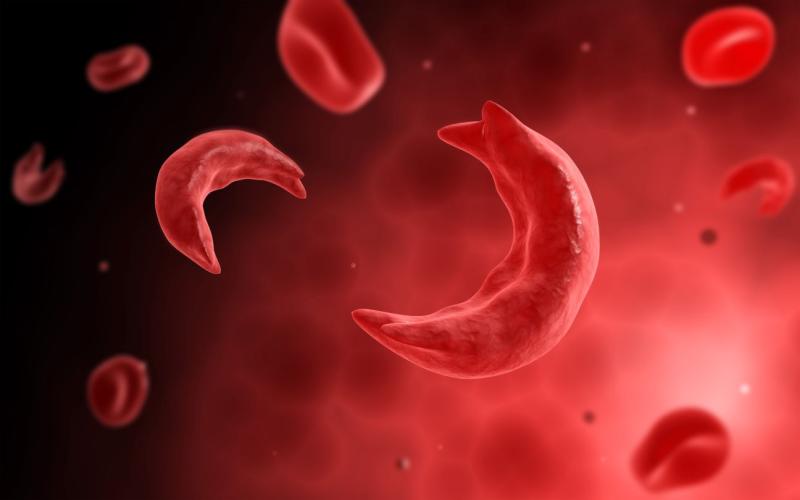
Sickle cell disease is a typical genetic hemoglobinopathy triggered by a point adaptation in beta-globin that stimulates the polymerization of deoxygenated haemoglobin resulting in red cell contortion, hemolytic anaemia, ischemic tissue deterioration, and microvascular blockage. Sickle cell anaemia is described as an intense hereditary structure of anaemia due to sickle cell disease where the mutated structure of haemoglobin distorts the red blood cell to become a crescent form at decreased oxygen level. Sickle cell disease possesses a class of pathological embodiment, whereas sickle cell anaemia is described as one such pathological embodiment of the sickle cell ailment. This description appears as the significant disparity between sickle cell disease and sickle cell anaemia.
What is Sickle Cell Disease?
Sickle cell disease is described as a regular genetic hemoglobinopathy triggered by a point transformation in beta-globin that enhances the polymerization of deoxygenated haemoglobin resulting in red cell contortion, ischemic tissue destruction, hemolytic anaemia, and microvascular blockage. Haemoglobin possesses a tetrameric form which is consisted of two tandems of alpha and beta chains. Typical adult red cells possess HbA ( a² and B²) as their permanent haemoglobin structure. In sickle cell disease, the glutamate residue in the 6th colon of the beta-globin gene is superseded by Valin. Adding to the HbA, individuals going through the pains of sickle cell disease possess a unique kind of haemoglobin in their red cells, known as the sickle haemoglobin acronym HbS. This alteration results in different structural and useful modifications in the haemoglobin molecule.
The Pathogenesis of Sickle Cell Disease
The voluntary inflow of cytosol of the red blood cell modifies into a dense gel when the partial force of oxygen declines below a particularly crucial stage. This is the pathological ground for the vital embodiment, which may include severe hemolysis, tissue destruction, and microvascular occlusion. Using continued deoxygenation, the HbS molecule polymerizes into long fibres within the red blood cell, deforming them to become crescent forms. The polymers begin to herniate using the red cell membrane when they mature. These structural changes of the red blood cell generate an inflow of Ca²+. Elevated intracellular calcium status then generates the cross-connection of the intracellular proteins, leading to an outflow of water and K+. The Reoccurrence of this procedure dehydrates the red blood cell, causing them to be tough and thick. Eventually, they become irreversibly sickled cells and are immediately removed from the spread by extravascular hemolysis. There are many ideas concerning the pathological motivation of microvascular occlusion, though the specific mechanism is unknown.
Medical Traits of Sickle Cell Disease
Sickle cell disease possesses an extensive range of clinical embodiment. Many influenced persons may lose their walking ability, while some have only mild indications. Sickle cell disease can be diagnosed using dithionate examination and haemoglobin electrophoresis to ascertain the availability of HbS. Prenatal diagnosis is conceivable by the analysis of fetal DNA acquired by amniocentesis.
What is Sickle Cell Anaemia?
The intense genetic structure of anaemia due to sickle cell disease, where the mutated structure of haemoglobin changes the red blood cell into a crescent form at reduced oxygen status, is described as sickle cell anaemia.
The Clinical Traits of Sickle Cell Anaemia
The clinical traits are intense hemolytic anaemia led by difficulties. There are four primary kinds of problems. These include:
- Vaso occlusive difficulty: these problems are more regular than the others and are predisposed by characteristics that have to do with deoxygenation, dehydration, acidosis, and infection. Due to the closure of the blood vessel, blood reserve to specific locations of the body, mainly to the extremities, comes to terms. Accordingly, infarct shows up in these locations, increasing severe pain. There is a disorder described as the hand-foot syndrome whereby the patient reports intense pain in the extremities.
- Visceral sequestration difficulty: this problem occurs due to the sickling and the pooling of blood within the organs. Sickle anaemia exacerbates an intense status during a visceral sequestration difficulty. Acute chest syndrome is the extensively hazardous intricacy of this difficulty.
- Aplastic difficulty: these occur as a result of parvovirus infection and, most times, due to folate depletion. Aplastic difficulties are featured by the quick drop in haemoglobin status, usually needing a transfusion.
- Hemolytic anaemia.
Management For Sickle Cell Anaemia
- Proper nutrition and hygiene
- Difficulties should be managed based on the patient’s situation, age, and drug submission.
- Folic acid
- Preventing the facets which spray the condition.
- Pneumococcal and meningococcal vaccination
Difference Between Sickle Cell Disease and Sickle Cell Anaemia
Sickle cell disease possesses different pathological embodiment. In contrast, sickle cell anaemia is a pathological embodiment of sickle cell disease.
Sickle cell disease is described as a regular genetic hemoglobinopathy triggered by a point modification in beta-globin that enhances the polymerization of deoxygenated haemoglobin resulting in red cell contortions, ischemic tissue destruction, and microvascular blockage. In contrast, sickle cell anaemia is an intense genetic structure due to sickle cell disease where the mutated structure of haemoglobin changes the red blood cells to possess a crescent form at reduced oxygen status.